| ELOCTATE® | Prescribing Information |
| [Antihemophilic factor (recombinant), Fc fusion protein], Lyophilized powder for solution for intravenous injection |
Rx only |
|
|||||||||||||||||||||||
BACK TO TOP
FULL PRESCRIBING INFORMATIONBACK TO TOP
1 INDICATIONS AND USAGE
ELOCTATE, Antihemophilic Factor (Recombinant), Fc Fusion Protein, is a recombinant DNA derived, antihemophilic factor indicated in adults and children with Hemophilia A (congenital Factor VIII deficiency) for:
- On-demand treatment and control of bleeding episodes,
- Perioperative management of bleeding,
- Routine prophylaxis to reduce the frequency of bleeding episodes.
Limitation of Use
ELOCTATE is not indicated for the treatment of von Willebrand disease.
2 DOSAGE AND ADMINISTRATION
For intravenous use after reconstitution only.
2.1 Dose
- Dose and duration of treatment depend on the severity of the Factor VIII deficiency, the location and extent of bleeding, and the patient's clinical condition. Careful monitoring of replacement therapy is necessary in cases of major surgery or life-threatening bleeding episodes.
- Each vial label of ELOCTATE states the Factor VIII potency in international units (IU). One IU corresponds to the activity of Factor VIII contained in one milliliter of normal human plasma.
- Potency assignment is determined using a chromogenic substrate assay. A field study1 has indicated that plasma Factor VIII levels can be monitored using either a chromogenic substrate assay or a one stage clotting assay routinely used in US clinical laboratories.
- Calculation of the required dose of Factor VIII is based on the empirical finding that 1 IU of Factor VIII per kg body weight raises the plasma Factor VIII level by 2 IU/dL.
The expected in vivo peak increase in Factor VIII level expressed as IU/dL (or % of normal) is estimated using the following formula:
Estimated Increment of Factor VIII (IU/dL or % of normal) = [Total Dose (IU)/body weight (kg)] × 2 (IU/dL per IU/kg)
The dose to achieve a desired in vivo peak increase in Factor VIII level may be calculated using the following formula:
Dose (IU) = body weight (kg) × Desired Factor VIII Rise (IU/dL or % of normal) × 0.5 (IU/kg per IU/dL)
- Patients may vary in their pharmacokinetic (e.g., half-life, in vivo recovery) and clinical responses. Base the dose and frequency of ELOCTATE on the individual clinical response.
- Dose adjustment may be necessary in pediatric patients under six years of age [see Use in Specific Populations (8.4)]. For patients six years of age or older, dose adjustment is not usually required.
On-demand Treatment and Control of Bleeding Episodes
A guide for dosing ELOCTATE for the on-demand treatment and control of bleeding episodes is provided in Table 1. Consideration should be given to maintaining a Factor VIII activity at or above the target range.
| Type of Bleeding | Factor VIII Level Required (IU/dL or % of normal) |
Dose (IU/kg) |
Frequency of Dosing (hours) | Duration of Therapy (days) |
|---|---|---|---|---|
| Minor and Moderate Joint, superficial muscle/no neurovascular compromise (except iliopsoas), deep laceration and renal, superficial soft tissue, mucous membranes |
40-60 | 20-30 | Repeat every 24-48 hours (12 to 24 hours for patients less than 6 years of age) |
Until the bleeding episode is resolved |
| Major Life or limb threatening hemorrhage, iliopsoas and deep muscle with neurovascular injury, retroperitoneum, intracranial, or gastrointestinal |
80-100 | 40-50 | Repeat every 12-24 hours (8 to 24 hours for patients less than 6 years of age) | Until bleeding is resolved (approximately 7-10 days) |
Perioperative Management
A guide for dosing ELOCTATE during surgery (perioperative management) is provided in Table 2. Consideration should be given to maintaining a Factor VIII activity at or above the target range.
| Type of Surgery | Factor VIII Level Required (IU/dL or % of normal) |
Dose (IU/kg) |
Frequency of Dosing (hours) |
Duration of Therapy (days) |
|---|---|---|---|---|
| Minor Uncomplicated tooth extraction |
50-80 | 25-40 | Repeat every 24 hours (12-24 hours for patients less than 6 years of age) | At least 1 day until healing is achieved |
| Major Intracranial, intra-abdominal, or joint replacement surgery |
80-120 (pre and post- operative) |
Preoperative: 40-60 Repeat: 40-50 |
Preoperative dose of 40 to 60 IU/kg followed by a repeat dose of 40-50 IU/kg after 8-24 hours (6 to 24 for patients less than 6 years of age) and then every 24 hours to maintain FVIII activity within the target range | Until adequate wound healing, then continue therapy for at least 7 days to maintain a Factor VIII activity within the target range |
Routine Prophylaxis
- The recommended starting regimen is 50 IU/kg of ELOCTATE administered every 4 days. Adjust the regimen based on patient response with dosing in the range of 25-65 IU/kg at 3-5 day intervals.
- For children <6 years of age, the recommended starting regimen is 50 IU/kg of ELOCTATE administered twice weekly. Adjust the regimen based on patient response with dosing in the range of 25-65 IU/kg at 3-5 day intervals. More frequent or higher doses up to 80 IU/kg may be required [see Use In Specific Populations (8.4), Clinical Pharmacology (12.3)].
2.2 Preparation and Reconstitution
- Use aseptic technique (clean and germ free) and a flat work surface during the reconstitution procedure.
- Allow the vial of ELOCTATE, containing the white to off-white lyophilized powder, and the pre-filled diluent syringe to reach room temperature before use.
- Remove the plastic cap from the vial and wipe the rubber stopper of the vial with an alcohol wipe. Allow the rubber stopper to dry.
-
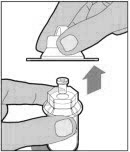
Completely remove the backing from the vial adapter package by peeling back the lid. Do not remove the vial adapter from the package or touch the inside of the package of the adapter.
-
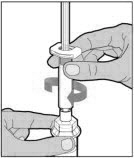
Place the vial on a flat and solid surface and use one hand to hold the vial steady. Use the other hand to place the vial adapter over the vial. Place the adapter spike directly above the center of the rubber stopper and push the adapter straight down until the spike punctures the center of the vial stopper and is fully inserted.
-
Lift the package cover away from the vial adapter and discard the cover.
-
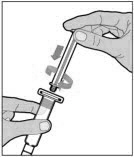
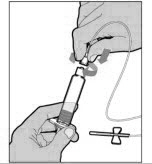
Hold the plunger rod at the circular disk. Place the tip of the plunger rod into the end of the syringe. Turn clockwise until it is securely attached. Only use the diluent syringe provided in the ELOCTATE package.
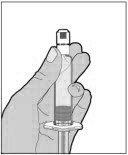
- With one hand, hold the diluent syringe by the ridged part directly under the cap, with the cap pointing up. Do not use if the cap has been removed or is not securely attached.
- With your other hand, grasp the cap and bend it at a 90° angle until it snaps off. After the cap snaps off, you will see the glass tip of the syringe. Do not touch the glass tip of the syringe or the inside of the cap.
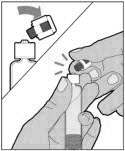
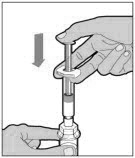
- With the vial sitting on a flat surface, insert the tip of the syringe into the adapter opening. Turn the syringe clockwise until it is securely attached to the adapter.
- Slowly depress the plunger rod to inject all of the diluent into the vial. The plunger rod may rise slightly after this process. This is normal.
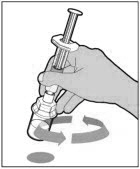
- With the syringe still connected to the adapter, gently swirl the vial until the product is completely dissolved. Do not shake. The reconstituted solution should be clear to slightly opalescent and colorless. Do not use the reconstituted ELOCTATE if it contains visible particles or is cloudy.
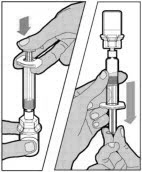
- Make sure the plunger rod is completely depressed. Turn the vial upside-down. Slowly pull on the plunger rod to draw the solution into the syringe. Be careful not to pull the plunger rod completely out of the syringe.
- Gently unscrew the syringe from the vial adapter and dispose of the vial with the adapter still attached. Do not touch the syringe tip or the inside of the cap.
- Use the reconstituted ELOCTATE as soon as possible, but no later than 3 hours after reconstitution. Do not touch the glass tip of the syringe if not used immediately after reconstitution. Protect from direct sunlight. Do not refrigerate after reconstitution.
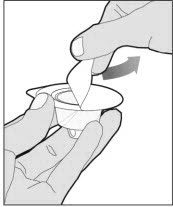
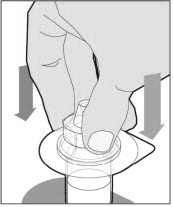
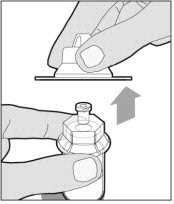
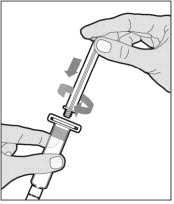
To combine two or more vials of ELOCTATE, after step 12 above, follow these pooling steps:
- Remove the diluent syringe from the vial adapter by turning it counterclockwise until it is completely detached.
- Leave the vial adapter attached to the vial, as it is needed for attaching a large luer lock syringe (not included in kit). Do not detach the diluent syringe until ready to attach the large luer-lock syringe.
- Attach a separate, large luer-lock syringe by turning clockwise until it is securely in place.
- Slowly pull on the plunger rod to draw the solution into the syringe.
- Repeat this pooling procedure with each vial that is needed to obtain the required dose. When pooling, do not detach the large luer-lock syringe until ready to attach it to the next vial (with vial adapter attached). Once you have pooled the required dose, proceed to administration using the large luer-lock syringe.
2.3 Administration
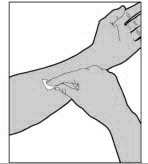
For intravenous injection only
- Inspect the reconstituted ELOCTATE solution visually for particulate matter and discoloration prior to administration. Do not use if particulate matter or discoloration is observed.
- Do not administer reconstituted ELOCTATE in the same tubing or container with other medications.
Administration Steps:
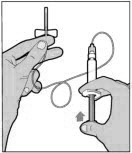
- Attach the syringe to the connector end of the infusion set tubing by turning clockwise until it is securely in place.
- Depress the plunger until all air is removed from the syringe and ELOCTATE has reached the end of the infusion set tubing. Do not push ELOCTATE solution through the needle.
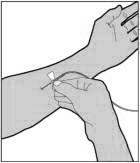
- Remove the protective needle cover from the infusion set tubing.
- Perform intravenous bolus infusion. The rate of administration should be determined by the patient's comfort level, and no faster than 10 ml per minute. After infusing ELOCTATE, remove and properly discard the infusion set.
3 DOSAGE FORMS AND STRENGTHS
ELOCTATE is available as a white to off-white lyophilized powder in single-dose vials containing nominally 250, 500, 750, 1000, 1500, 2000, 3000, 4000, 5000 or 6000 international units (IU) per vial. The actual Factor VIII potency is labeled on each ELOCTATE vial.
4 CONTRAINDICATIONS
ELOCTATE is contraindicated in patients who have had life-threatening hypersensitivity reactions to ELOCTATE or its excipients (sucrose, sodium chloride, L-histidine, calcium chloride and polysorbate 20).
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions have been reported with ELOCTATE. Allergic-type hypersensitivity reactions, including anaphylaxis, have been reported with Factor VIII replacement products. Early signs of hypersensitivity reactions that can progress to anaphylaxis may include angioedema, chest tightness, dyspnea, wheezing, urticaria, and pruritus. Immediately discontinue administration and initiate appropriate treatment if hypersensitivity reactions occur.
5.2 Neutralizing Antibodies
Formation of neutralizing antibodies (inhibitors) to Factor VIII has been reported following administration of ELOCTATE. Monitor all patients for the development of Factor VIII inhibitors by appropriate clinical observations and laboratory tests. If the plasma Factor VIII level fails to increase as expected or if bleeding is not controlled after ELOCTATE administration, suspect the presence of an inhibitor (neutralizing antibody) [see Warnings and Precautions (5.5)].
5.3 Cardiovascular Risk Factors
Hemophilic patients with cardiovascular risk factors or diseases may be at the same risk to develop cardiovascular events as non-hemophilic patients when clotting has been normalized by treatment with Factor VIII.
5.5 Monitoring Laboratory Tests
Monitor plasma Factor VIII activity by performing a validated test (e.g., one stage clotting assay), to confirm that adequate Factor VIII levels have been achieved and maintained [see Dosage and Administration (2)].
Monitor for the development of Factor VIII inhibitors. Perform a Bethesda inhibitor assay if expected Factor VIII plasma levels are not attained, or if bleeding is not controlled with the expected dose of ELOCTATE. Use Bethesda Units (BU) to report inhibitor levels.
6 ADVERSE REACTIONS
The most frequently occurring adverse reactions (incidence >0.5% of subjects) reported in previously treated patients (PTPs) clinical trials were arthralgia, malaise, myalgia, headache, and rash. The most frequently occurring adverse reactions (incidence ≥1.0% of subjects) reported in previously untreated patients (PUPs) clinical trials were Factor VIII inhibition, device-related thrombosis, and rash papular.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of one drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice.
Previously Treated Patients (PTPs)
ELOCTATE has been evaluated in 276 subjects in five completed studies in previously treated patients (PTPs) with severe Hemophilia A (<1% endogenous FVIII activity or a genetic mutation consistent with severe Hemophilia A) who received at least one dose of ELOCTATE as part of either routine prophylaxis, on-demand treatment of bleeding episodes or perioperative management. Sixty-nine (25.0%) were pediatric subjects <12 years of age, 25 (9.1%) were adolescents (12 to <18 years of age), and 182 (65.9%) were adults (18 years of age and older). There were 200 subjects treated for at least 104 weeks, 151 subjects treated for at least 156 weeks and 107 subjects treated for at least 208 weeks. The total number of exposure days (EDs) was 80,848 with a median of 294 (range 1-735) exposure days per subject. The subjects received a total of 82,024 injections with a median of 303.5 injections of ELOCTATE (range 1-755) per subject. Adverse events (AEs) were monitored for a total of 893.72 subject-years. Two subjects (0.7% of total 276) with cardiovascular risk factors each experienced a serious adverse reaction of myocardial infarction during the study.
Adverse reactions (ARs) were reported for 11 of 276 (4.0%) subjects treated with routine prophylaxis or episodic (on-demand) therapy. No age-specific differences in ARs were observed between pediatric and adult subjects. Table 3 summarizes the most frequently occurring adverse reactions in PTPs. Additional adverse reactions, each occurring in a single subject (incidence 0.4%), include dizziness, dysgeusia, bradycardia, hypertension, hot flush, angiopathy (investigator term: vascular pain after injection of study drug), cough, lower abdominal pain, back pain, joint swelling, chest pain, feeling cold, feeling hot, and procedural hypotension. Two subjects were withdrawn from study due to adverse reactions of rash and arthralgia. In the studies, no inhibitors were detected and no events of anaphylaxis were reported.
| MedDRA* System Organ Class | Adverse Reactions | Number of Subjects n (%) |
|---|---|---|
|
||
| Nervous system disorders | Headache | 2 (0.7) |
| Skin and subcutaneous tissue disorders | Rash | 2 (0.7) |
| Musculoskeletal and connective tissue disorders | Arthralgia Myalgia |
2 (0.7) 2 (0.7) |
| General disorders and administration site conditions | Malaise |
2 (0.7) |
Previously Untreated Patients (PUPs)
ELOCTATE safety was also evaluated in one completed study (PUPs study) in 103 subjects with severe hemophilia A (<1% endogenous FVIII activity). Overall, the median number of weeks on treatment was 64.24 weeks (range: 0.0–206.8 weeks). The number of subjects with at least 10 exposure days (EDs) was 87 (84.5%), at least 20 EDs was 85 (82.5%), and at least 50 EDs was 81 (78.6%).
Adverse drug reactions (ADRs) were reported in 29 of 103 (28.2%) subjects treated with ELOCTATE on routine prophylaxis, episodic, and/or immune tolerance induction (ITI) therapies. ADRs in PUPs are summarized in Table 4.
| MedDRA* System Organ Class | Adverse Reactions | Number of Subjects (%) N=103† |
|---|---|---|
|
||
| Blood and lymphatic system disorders | Factor VIII inhibition‡ | 28 (27.2) |
| General disorders and administration site conditions | Device related thrombosis§ | 2 (1.9) |
| Skin and subcutaneous tissues disorders | Rash papular | 1 (1.0) |
Immunogenicity
Clinical trial subjects were monitored for neutralizing antibodies to Factor VIII. No PTPs developed confirmed neutralizing antibodies to Factor VIII. One 25 year old subject had a transient, positive, neutralizing antibody of 0.73 BU at week 14, which was not confirmed upon repeat testing 18 days later and thereafter.
In PUPs Study, development of neutralizing antibodies (inhibitors) was observed in 28 subjects, 14 of them had a high-titer inhibitor. Based on subjects with an inhibitor test following an exposure day (ED) milestone or who developed an inhibitor at any time during the study, the incidence of Factor VIII inhibitor development was:
- 28/90 subjects (31.1%) with at least 10 EDs
- 28/86 subjects (32.6%) with at least 50 EDs
The median time to inhibitor development for the 28 subjects was 9 EDs (interquartile range: 6.5–12).
The detection of antibodies that are reactive to Factor VIII is highly dependent on many factors, including the sensitivity and specificity of the assay, sample handling, timing of sample collection, concomitant medications and underlying disease. Therefore, it may be misleading to compare of the incidence of antibodies to ELOCTATE with the incidence of antibodies to other products.
6.2 Postmarketing Experience
The following adverse reactions have been identified during the postapproval use of ELOCTATE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: Factor VIII inhibitor development
Immune system disorders: hypersensitivity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no studies of ELOCTATE use in pregnant women to inform a drug-associated risk. The background risk of major birth defects and miscarriage in the indicated population is unknown; however, the background risk of major birth defects in the U.S. general population is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies.
Animal reproductive and developmental toxicity studies have not been conducted with ELOCTATE. In a placental transfer study, ELOCTATE was detected in murine fetal blood samples at approximately 1% of the maternal blood levels (range, 0.2% to 1.9%), 3 to 4 hours following dosing of pregnant mice with 260 to 650 times the clinical dose of 20 to 50 IU/kg ELOCTATE [see Data].
It is not known whether ELOCTATE can cause fetal harm when administered to a pregnant woman, or whether it can affect reproduction capacity. If ELOCTATE is clearly needed to treat a pregnant woman, advise the patient that the risks to the mother and to the fetus are unknown.
Animal data
Pregnant, genetically-modified, FVIII-deficient mice (Hem A mice) were injected intravenously with a single dose of 400 IU (approximately 13,000 IU/kg) ELOCTATE at the end of pregnancy on Gestation Day 19. Blood samples were collected from the maternal mice and the fetuses 3 to 4 hours after dosing, and FVIII activity was measured in both maternal and fetal plasma using a FVIII chromogenic assay. After dosing pregnant HemA mice with ELOCTATE, FVIII activity in fetal blood was approximately 1% of the maternal blood levels, suggesting that placental transfer of ELOCTATE may occur. The relevance of these data to humans is unknown.
8.2 Lactation
Risk Summary
There is no information regarding the presence of ELOCTATE in human milk, its effects on the breastfed infant, or its effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ELOCTATE and any potential adverse effects on the breastfed infant from ELOCTATE or from the underlying maternal condition.
8.4 Pediatric Use
Safety and efficacy studies have been performed in 82 previously treated, pediatric patients (PTPs) <18 years of age who received at least one dose of ELOCTATE as part of routine prophylaxis, on-demand treatment of bleeding episodes, or perioperative management. Adolescent subjects were enrolled in the adult and adolescent safety and efficacy trial, and subjects <12 were enrolled in a pediatric trial. Safety and efficacy of ELOCTATE have been evaluated in 103 previously untreated pediatric patients (PUPs) <6 years of age (median 0.58 year; range: 0.02–4 years) in one study (PUPs Study) [see Adverse Reactions (6)].
Pharmacokinetic data from a pediatric study of the 54 evaluable subjects <12 years of age showed that no dose adjustment was required for patients ≥6 years old. Children age 1 to 5 years had a shorter half-life and higher clearance (adjusted for body weight); therefore, a higher dose or more frequent dosing may be needed in this age group [see Clinical Pharmacology (12.3)].
11 DESCRIPTION
ELOCTATE, Antihemophilic Factor (Recombinant), Fc Fusion Protein, is a sterile, non-pyrogenic, white to off-white lyophilized powder for reconstitution for intravenous injection. The product is supplied in single-dose vials containing nominal potencies of 250, 500, 750, 1000, 1500, 2000 3000, 4000, 5000 or 6000 international units (IU). Each vial of ELOCTATE is labeled with the actual content in IU. The powder for injection is reconstituted with 3 mL sterile water for injection (SWFI) supplied in a sterile prefilled syringe. The reconstituted solution should be essentially free of particles. The final product contains the excipients: sucrose, sodium chloride, L-histidine, calcium chloride and polysorbate 20. ELOCTATE contains no preservatives.
B-domain deleted recombinant Factor VIII, Fc fusion protein (BDD-rFVIIIFc) is the active ingredient in ELOCTATE. BDD-rFVIIIFc is a recombinant protein consisting of a B-domain deleted analogue of human Coagulation Factor VIII covalently linked to the human immunoglobulin G1 (IgG1) Fc domain sequence. The Factor VIII portion of the molecule has a 90 kDa heavy chain and an 80 kDa light chain (similar to endogenous Factor VIII), which are linked by 14 (of 908) amino acids from the central B-domain. The FVIII portion has post-translational modifications comparable to endogenous Factor VIII. The Fc domain of the molecule contains the hinge, CH2, and CH3 regions of IgG1. BDD-rFVIIIFc contains 1890 amino acids with an apparent molecular weight of 220 kDa. The majority of the expressed protein is proteolytically processed to a two chain molecule; however, ELOCTATE may also contain up to 39% of a single chain, non-processed form. Both molecules have been shown to have comparable Factor VIII activity.
BDD-rFVIIIFc is produced by recombinant DNA technology from a human embryonic kidney (HEK) cell line, which has been extensively characterized. The HEK cell line expresses BDD-rFVIIIFc into a defined, cell culture medium that does not contain any proteins derived from animal or human sources. BDD-rFVIIIFc is purified using a series of chromatography steps, including affinity capture with a recombinant, single chain antibody fragment produced in a yeast expression system. No human or animal derived proteins are used in the purification or formulation processes. The production process also incorporates two dedicated viral clearance steps - a detergent treatment step for inactivation and a 15 nm filtration step for removal of viruses.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
ELOCTATE is a recombinant fusion protein that temporarily replaces the missing Coagulation Factor VIII needed for effective hemostasis. ELOCTATE contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life.
12.2 Pharmacodynamics
Hemophilia A is a bleeding disorder characterized by a deficiency of functional coagulation Factor VIII, resulting in a prolonged, patient plasma clotting time as measured by the activated partial thromboplastin time (aPTT) assay. Treatment with ELOCTATE normalizes the aPTT over the effective dosing period.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of ELOCTATE (rFVIIIFc) were evaluated in 28 subjects following a 10 minute intravenous infusion of a single dose of 50 IU/kg. The PK parameters were based on plasma FVIII activity measured by the one-stage clotting assay. The PK profile obtained at week 14, after repeated dosing, was comparable with the PK profile obtained after the first dose. The PK data demonstrate that ELOCTATE has a prolonged circulating half-life. Time to 1% was 5.10 days (95% CI: 4.54, 5.66). The terminal plasma half-life of ELOCTATE when compared against a currently marketed recombinant Factor VIII (ADVATE®) was 1.5 fold longer.
Pediatric and Adolescent Pharmacokinetics
Pharmacokinetic (PK) parameters of ELOCTATE were determined for adolescents (ages 12 to 17 years) in the adult and adolescent study and for children (ages 1 to 5 years and 6 to 11 years) in the pediatric study. Table 5 presents the PK parameters calculated from the pediatric data of 65 subjects, less than 18 years of age, after receiving a single 50 IU/kg dose.
Compared to adults and adolescents, body weight adjusted clearance was 75% higher in children 1 to 5 years of age. These results indicate a need for dose adjustments in children 1 to 5 years of age.
The PK evaluation of pediatric subjects, ages 6 to 17 years, showed that their PK profiles and arithmetic means of PK parameters are similar to those of adults. Therefore, for subjects 6 years and older, an age-based dose adjustment is not required.
| PK Parameters* | Pediatric Study | Adult and Adolescent Study | ||
|---|---|---|---|---|
| 1 to 5 Years | 6 to 11 Years | 12 to 17 Years | Adults† | |
| N=23 | N=31 | N=11 | N=28 | |
|
Abbreviations: CI = confidence interval; AUC = area under the FVIII activity time curve; t1/2 = terminal half-life; MRT = mean residence time; CL = body weight adjusted clearance; Vss = body weight adjusted volume of distribution at steady state |
||||
| Incremental Recovery (IU/dL per IU/kg) |
1.92 (1.80, 2.04) |
2.44 (2.07, 2.80) |
1.85 (1.58, 2.12) |
2.26 (2.13, 2.40) |
| AUC/Dose (IU x h/dL per IU/kg) |
30.0 (26.5, 33.6) |
41.9 (34.0, 49.8) |
38.7 (34.3, 43.1) |
54.1 (47.0, 61.1) |
| t½ (h) | 12.7 (11.2, 14.1) |
14.9 (12.0, 17.8) |
16.4 (14.1, 18.6) |
19.7 (17.4, 22.0) |
| MRT (h) | 17.2 (15.4, 19.1) |
20.9 (17.1, 24.7) |
23.1 (19.9, 26.4) |
26.1 (23.2, 28.9) |
| CL (mL/h/kg) | 3.60 (3.13, 4.07) |
2.78 (2.44, 3.13) |
2.66 (2.34, 2.98) |
2.06 (1.78, 2.34) |
| Vss (mL/kg) | 58.6 (54.9, 62.3) |
52.1 (45.3, 59.0) |
60.3 (53.3, 67.3) |
49.5 (46.9, 52.2) |
BACK TO TOP
14 CLINICAL STUDIES
The safety and efficacy of ELOCTATE was evaluated in two multicenter, prospective, open-label clinical trials in previously treated patients (PTPs) (adult and adolescent study and pediatric study), and in an extension study.
The adult and adolescent study compared the efficacy of each of two prophylactic treatment regimens (individualized and fixed weekly) to episodic (on-demand) treatment; determined hemostatic efficacy in the treatment of bleeding episodes; and determined hemostatic efficacy during perioperative management in subjects undergoing major surgical procedures. The study enrolled a total of 165 previously treated male patients (PTPs) with severe Hemophilia A (<1% endogenous Factor VIII activity or a genetic mutation consistent with severe Hemophilia A). Subjects were aged 12 to 65 years, including 13 pediatric subjects aged 12 to 17 years. Of the 165 enrolled subjects, 164 received at least one dose of ELOCTATE and 163 (98%) were evaluable for efficacy. A total of 153 subjects (93%) completed the study.
The pediatric study evaluated the efficacy of individualized prophylactic treatment; determined hemostatic efficacy in the treatment of bleeding episodes; and determined hemostatic efficacy during perioperative management in subjects undergoing surgical procedures. The study enrolled a total of 71 previously treated male pediatric patients with severe hemophilia A (<1% endogenous FVIII activity or a genetic mutation consistent with severe hemophilia A). Of the 71 enrolled subjects, 69 received at least 1 dose of ELOCTATE and were evaluable for efficacy. All subjects were less than 12 years of age (35 were 1 to 5 years of age and 34 were 6 to 11 years of age).
The extension study evaluated the safety and efficacy of prophylactic treatment regimens or on-demand treatment; as well as hemostatic efficacy during perioperative management in subjects undergoing surgical procedures. The study enrolled a total of 240 previously treated male patients (aged 2 to 66 years old) with severe hemophilia A who completed the adult and adolescent study or the pediatric study.
On-demand Treatment and Control of Bleeding Episodes
In the adult and adolescent study, a total of 757 bleeding episodes in 106 subjects were treated with ELOCTATE. The majority of the bleeding episodes were spontaneous and localized in joints. The median dose per injection used to treat a bleeding episode was 27.35 (IQR 22.73, 32.71) IU/kg. Assessment of response to each injection was recorded by subjects at 8-12 hours after treatment. A 4-point rating scale of excellent, good, moderate, and no response was used to assess response. Efficacy in control of bleeding episodes in subjects ≥12 years of age is summarized in Table 6.
| New bleeding episodes | (n=757) | |
|---|---|---|
|
||
| # of Injections to treat bleeding episodes | ||
| 1 injection | 661 (87.3%) | |
| 2 injections | 79 (10.4%) | |
| >2 injections | 17 (2.2%) | |
| Response to first injection* | (n=745) | |
| Excellent or good | 78.1% | |
| Moderate | 21.2% | |
| No response | 0.7% | |
In the pediatric study, a total of 86 bleeding episodes in 69 pediatric subjects were treated with ELOCTATE. Assessment of response to each injection was recorded by subjects at 8 to 12 hours post-treatment. A 4-point rating scale of excellent, good, moderate, and no response was used to assess response. Efficacy in control of bleeding episodes in subjects <12 years of age is summarized in Table 7.
The hemostatic efficacy in treatment of bleeds was rated excellent or good in 92.6% for all evaluable first injections.
| 1-5 Years (n=35) |
6 to 11 Years (n=34) |
Total (<12 Years) (n=69) |
||
|---|---|---|---|---|
|
||||
| New bleeding episodes | (n=38) | (n=48) | (n=86) | |
| # of Injections to treat bleeding episodes | 1 injection | 29 (76.3%) | 41 (85.4%) | 70 (81.4%) |
| 2 injections | 7 (18.4%) | 3 (6.3%) | 10 (11.6%) | |
| >2 injections | 2 (5.3%) | 4 (8.3%) | 6 (7.0%) | |
| Median dose per injection (IU/kg) to treat a bleeding episode (IQR) | 51.35 (29.94, 59.52) |
48.15 (29.08, 55.97) |
49.69 (29.41, 56.82) |
|
| Median total dose (IU/kg) to treat a bleeding episode (IQR) | 56.40 (29.94, 72.46) |
53.49 (29.08, 66.80) |
54.90 (29.41, 71.09) |
|
| Response to first injection* | (n=35) | (n=46) | (n=81) | |
| Excellent or good | 32 (91.4%) | 43 (93.5%) | 75 (92.6%) | |
| Moderate | 3 (8.6%) | 1 (2.2%) | 4 (4.9%) | |
| No response | 0 (0.0%) | 2 (4.3%) | 2 (2.5%) | |
Major surgeries
Hemostasis was assessed in forty-five (45) surgeries in thirty-two (32) subjects from the adult and adolescent study and the extension study. There were no major surgeries in the pediatric study or the pharmacokinetics studies. Of the 45 major surgeries, 36 surgeries (80.0%) required a single perioperative dose to maintain hemostasis. Of the 42 major surgeries treated with at least one dose, the median average dose per injection to maintain hemostasis during surgery was 59.1 IU/kg (range 35-111). On the day of surgery, most subjects received a second injection. The total dose on the day of surgery ranged from 37.6-157.9 IU/kg.
Hemostatic response was assessed by the investigator using ordinal scales as follows:
Excellent: Intraoperative and postoperative blood loss similar to (or less than) the nonhemophilic patient. No extra doses of rFVIIIFc needed and blood component transfusions required are similar to nonhemophilic patient
Good: Intraoperative and/or postoperative bleeding slightly increased over expectations for the nonhemophilic patient, but the difference was not clinically significant. Intraoperative blood loss no more than 250 mL greater than expected for a nonhemophilic patient and no extra doses of rFVIIIFc needed and blood component transfusions required are similar to nonhemophilic patient
Fair: Intraoperative and/or postoperative blood loss is increased over expectation for the nonhemophilic patient and additional treatment was needed. Intraoperative blood loss 250 to 500 mL greater than expected for person without hemophilia or extra dose of rFVIIIFc needed or increased blood component transfusion requirement
Poor/none: Significant intraoperative and/or postoperative bleeding that was substantially increased over expectations for the nonhemophilic patient, required intervention, and was not explained by a surgical/medical issue other than hemophilia: Intraoperative blood loss >500 mL greater than for the nonhemophilic patient or unexpected hypotension or unexpected transfer to intensive care unit due to bleeding or substantially increased blood component transfusion requirement.
For forty-one (41) major surgeries in twenty-nine (29) subjects, hemostatic response was assessed and rated as excellent in 38 (93%) surgeries and good in 3 (7%) surgeries.
Types of surgeries assessed include major orthopedic procedures such as joint replacements (bilateral knee, as well as unilateral elbow, hip and knee replacements), ankle fusion and amputation. Other major surgeries include appendectomy, arthroscopy, spinal surgery, and inguinal hernia repair.
Minor surgeries
A hemostatic assessment of 72 minor surgical procedures in 59 subjects from all three studies was conducted with all (100%) having excellent or good response (excellent response [61 of 72; 84.7%] and good response [11 of 72; 15.3%]).
Adult and adolescent study
The efficacy of routine prophylaxis was evaluated against on-demand treatment. A total of 117 subjects received an individualized, twice weekly regimen, which started with 25 IU/kg on the first day followed by 50 IU/kg on the fourth day. The dose and interval were adjusted within the range of 25-65 IU/kg every 3-5 days to maintain trough levels between 1% and 3% above baseline, or higher, as clinically indicated to prevent bleeding. The median dosing interval was 3.5 days. Among the 112 subjects treated for at least 6 months, 111 (99%) achieved a dosing interval of three days or longer, 39 (35%) achieved a dosing interval of 4 days or longer, and 33 (29%) achieved a dosing interval of 5 days or longer during the last 3 months on study. Twenty-three subjects received 65 IU/kg of ELOCTATE once weekly for a median period of 28 weeks. An additional 23 subjects received episodic (on-demand) doses of ELOCTATE for the treatment of bleeding episodes and were on study for a median period of 29 weeks. Using a negative binomial model to analyze the annualized bleeding rate (ABR), there was a statistically significant reduction in ABR of 92% (p<0.001) for subjects in the individualized prophylaxis arm and a statistically significant reduction of 76% (p<0.001) for subjects in the weekly prophylaxis arm compared to the episodic (on-demand) arm. Fifty-three (53) of 117 (45%) subjects experienced no bleeding episodes while on individualized prophylaxis and 4 of 23 (17%) subjects experienced no bleeding episodes while on weekly prophylaxis.
Median ABRs in subjects evaluable for efficacy is summarized in Table 8.
| Bleeding Episode Etiology | Individualized Prophylaxis
(N=117) |
Episodic (On-Demand) (N=23) |
|---|---|---|
|
||
| Overall ABR | 1.6 | 33.6 |
| (0.0, 4.7) | (21.1, 48.7) | |
| Spontaneous ABR | 0.0 | 20.2 |
| (0.0, 2.0) | (12.2, 36.8) | |
| Joint ABR | 0.0 | 22.8 |
| (0.0, 3.1) | (15.1, 39.0) | |
Pediatric study
Sixty-nine (69) subjects received ELOCTATE on an individualized prophylactic dose regimen starting with a twice weekly regimen consisting of 25 IU/kg on the first day followed by 50 IU/kg on the fourth day. The dose could be adjusted within the range of 25-80 IU/kg with a minimum dosing interval of every 2 days to maintain trough of 1% above baseline or as clinically indicated to prevent bleeding. The median dosing interval was 3.49 days (interquartile range, 3.46 to 3.51 days) with no difference in the median dosing interval between age cohorts. 89.9% of subjects remained on a twice weekly interval. The median weekly dose of ELOCTATE for subjects 1-5 years of age was 91.63 IU/kg (interquartile range (IQR), 84.72 to 104.56 IU/kg). For subjects in the 6 to 11 years of age cohort, the median weekly dose was 86.88 IU/kg (IQR, 79.12 to 103.08 IU/kg).
Of all subjects, 32 (46.4%) experienced no bleeding episodes (18 subjects (51.4%) 1-5 years of age and 14 subjects (41.2%) 6 to 11 years of age). A presentation of the median ABRs evaluable for efficacy is summarized in Table 9.
| Bleeding Episode Etiology | 1-5 Years (N=35) |
6 to 11 Years (N=34) |
Total (<12 Years) (N=69) |
|---|---|---|---|
|
|||
| Overall ABR | 0.0 | 2.0 | 2.0 |
| (0.0, 4.0) | (0.0, 4.0) | (0.0, 4.0) | |
| Spontaneous ABR | 0.0 | 0.0 | 0.0 |
| (0.0, 0.0) | (0.0, 0.0) | (0.0, 0.0) | |
| Joint ABR | 0.0 | 0.0 | 0.0 |
| (0.0, 1.9) | (0.0, 2.0) | (0.0, 2.0) | |
15 REFERENCES
- Sommer JM, Moore N, McGuffie-Valentine B, et al. Comparative field study evaluating the activity of recombinant factor VIII Fc fusion protein in plasma samples at clinical haemostasis laboratories. Haemophilia. 2014;20:294–300.
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ELOCTATE is supplied in kits comprising a single-dose vial containing nominally, 250, 500, 750, 1000, 1500, 2000, 3000, 4000, 5000 or 6000 international units (IU) of Factor VIII potency, a pre-filled syringe with 3 mL sterile water for injection, and a sterile vial adapter (reconstitution device). The actual amount of ELOCTATE in IU is stated on the label and carton of each vial.
Not made with natural rubber latex.
| Strength | Potency Color Code | Kit NDC Number |
| 250 IU | Yellow | 71104-801-01 |
| 500 IU | Red | 71104-802-01 |
| 750 IU | Garnet | 71104-803-01 |
| 1000 IU | Green | 71104-804-01 |
| 1500 IU | Dark Green | 71104-805-01 |
| 2000 IU | Royal Blue | 71104-806-01 |
| 3000 IU | Mist Grey | 71104-807-01 |
| 4000 IU | Orange | 71104-808-01 |
| 5000 IU | Mist Brown | 71104-809-01 |
| 6000 IU | Purple | 71104-810-01 |
Prior to reconstitution:
- Store ELOCTATE in the original package to protect the ELOCTATE vials from light.
- Store ELOCTATE in powder form at 2°C to 8°C (36°F to 46°F). Do not freeze to avoid damage to the pre-filled diluent syringe.
- ELOCTATE may be stored at room temperature, not to exceed 30°C (86°F), for a single period of up to 6 months, within the expiration date printed on the label.
- If stored at room temperature, record the date that ELOCTATE is removed from refrigeration on the carton in the area provided. After storage at room temperature, do not return the product to the refrigerator.
- Do not use beyond the expiration date printed on the vial or 6 months after the date that was written on the carton, whichever is earlier.
After Reconstitution:
- The reconstituted product may be stored at room temperature, not to exceed 30°C (86°F), for up to 3 hours. Protect from direct sunlight. After reconstitution, if the product is not used within 3 hours, it must be discarded.
- Do not use ELOCTATE if the reconstituted solution is cloudy or has particulate matter.
- Discard any unused ELOCTATE.
17 PATIENT COUNSELING INFORMATION
Advise the patients to:
- Read the FDA approved patient labeling (Patient Information and Instructions for Use).
- Call their healthcare provider or go to the emergency department right away if a hypersensitivity reaction occurs. Early signs of hypersensitivity reactions may include rash, hives, itching, facial swelling, tightness of the chest, and wheezing.
- Contact their healthcare provider or treatment facility for further treatment and/or assessment if they experience a lack of a clinical response to Factor VIII therapy because this may be a sign of inhibitor development.
Manufactured by:
Bioverativ Therapeutics Inc.
Waltham, MA 02451 USA
U.S. License Number 2078
©2020 Bioverativ Therapeutics Inc. All rights reserved.
ELOCTATE® is a registered trademark of Bioverativ.
ADVATE® is a registered trademark of Baxalta Incorporated.
Patient Information
ELOCTATE® /el' ok' tate /
[Antihemophilic Factor (Recombinant), Fc Fusion Protein]
Please read this Patient Information carefully before using ELOCTATE and each time you get a refill, as there may be new information. This Patient Information does not take the place of talking with your healthcare provider about your medical condition or your treatment.
What is ELOCTATE?
ELOCTATE is an injectable medicine that is used to help control and prevent bleeding in people with Hemophilia A (congenital Factor VIII deficiency).
Your healthcare provider may give you ELOCTATE when you have surgery.
Who should not use ELOCTATE?
You should not use ELOCTATE if you had an allergic reaction to it in the past.
What should I tell my healthcare provider before using ELOCTATE?
Talk to your healthcare provider about:
- Any medical problems that you have or had.
- All prescription and non-prescription medicines that you take, including over-the-counter medicines, supplements or herbal medicines.
- Pregnancy or if you are planning to become pregnant. It is not known if ELOCTATE may harm your unborn baby.
- Breastfeeding. It is not known if ELOCTATE passes into the milk and if it can harm your baby.
How should I use ELOCTATE?
You get ELOCTATE as an infusion into your vein. Your healthcare provider will instruct you on how to do infusions on your own, and may watch you give yourself the first dose of ELOCTATE.
Contact your healthcare provider right away if bleeding is not controlled after using ELOCTATE.
What are the possible side effects of ELOCTATE?
You can have an allergic reaction to ELOCTATE. Call your healthcare provider or emergency department right away if you have any of the following symptoms: difficulty breathing, chest tightness, swelling of the face, rash or hives.
Your body can also make antibodies called "inhibitors" against ELOCTATE. This can stop ELOCTATE from working properly. Your healthcare provider may give you blood tests to check for inhibitors.
Additional common side effects of ELOCTATE are headache, rash, joint pain, muscle pain, and general discomfort.
If you have risk factors for developing abnormal blood clots in your body, such as an indwelling venous catheter, treatment with Factor VIII may increase this risk.
These are not the only possible side effects of ELOCTATE. Tell your healthcare provider about any side effect that bothers you or does not go away.
How should I store ELOCTATE?
- Keep ELOCTATE in its original package.
- Protect it from light.
- Do not freeze.
- Store refrigerated (2°C to 8°C or 36°F to 46°F) or at room temperature [not to exceed 30°C (86°F)], for up to six months.
- When storing at room temperature:
- -
- Note on the carton the date on which the product is removed from refrigeration.
- -
- Use the product before the end of this 6-month period or discard it.
- -
- Do not return the product to the refrigerator.
Do not use ELOCTATE after the expiration date printed on the vial or, if you removed it from the refrigerator, after the date that was noted on the carton, whichever is earlier.
After reconstitution (mixing with the diluent):
- Do not use ELOCTATE if the reconstituted solution is not clear to slightly opalescent and colorless.
- Use reconstituted product as soon as possible.
- You may store reconstituted solution at room temperature, not to exceed 30°C (86°F), for up to three hours. Protect the reconstituted product from direct sunlight. Discard any product not used within three hours.
What else should I know about ELOCTATE?
Medicines are sometimes prescribed for purposes other than those listed here. Do not use ELOCTATE for a condition for which it was not prescribed. Do not share ELOCTATE with other people, even if they have the same symptoms that you have.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Bioverativ Therapeutics Inc.
Waltham, MA 02451 USA
U.S. License Number 2078
©2020 Bioverativ Therapeutics Inc. All rights reserved.
ELOCTATE® is a registered trademark of Bioverativ.
Revised: December 2020
ELOCTATE®
Antihemophilic Factor (Recombinant), Fc Fusion Protein
INSTRUCTIONS FOR USE
Read the Instructions for Use before you start using ELOCTATE and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
Your healthcare provider should show you or your caregiver how to reconstitute and administer ELOCTATE the first time ELOCTATE is used.
Check the expiration date on the ELOCTATE kit.
Do not use the product if past the expiration date.
Allow the ELOCTATE vial and the diluent to come to room temperature.
Do not use external heat sources such as putting the vial and/or diluent in hot water.
Find a clean, flat work surface and collect all the supplies you will need to reconstitute and administer ELOCTATE.
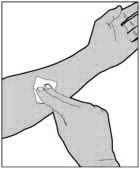
Wash your hands with soap and water. Aseptic technique (clean and germ free) should be used.
YOUR KIT CONTAINS:
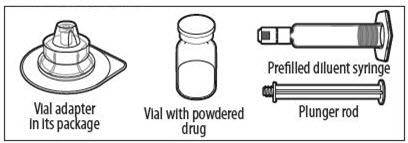
RECONSTITUTION
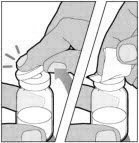
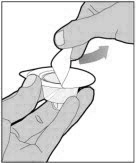
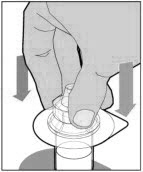

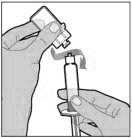


POOLING
POOLING is the process of combining two or more reconstituted vials into a larger syringe (not into the diluent syringe) prior to intravenous administration.
If you are using two or more vials, follow these pooling steps.
Be sure to leave the vial adapter attached to the vial as you will need it for attaching a large luer lock syringe.
Do not detach the diluent syringe or the large luer syringe until you are ready to attach the large luer lock syringe to the next vial (with vial adapter attached).
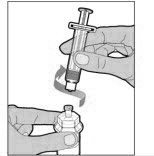
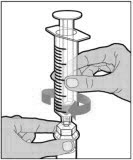
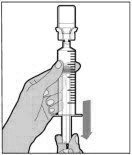
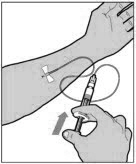
ADMINISTRATION (Intravenous Injection)
ELOCTATE is administered by intravenous infusion after reconstitution of the drug powder with the diluent.
Your healthcare provider should teach you how to infuse ELOCTATE. Once you have been taught to self-infuse, you can follow these instructions.
Do not administer reconstituted ELOCTATE if it contains particulate matter, is discolored, or is cloudy.
STORAGE CONDITIONS - PRODUCT KIT
Keep refrigerated until use.
Keep away from direct sunlight.
STORAGE CONDITIONS - RECONSTITUTED
ELOCTATE should be administered within 3 hours after reconstitution.
Do not refrigerate after reconstitution.
Keep away from direct sunlight.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured for:
Bioverativ Therapeutics Inc.
Waltham, MA 02451 USA
U.S. License Number 2078
©2014-2017 Bioverativ Therapeutics Inc. All rights reserved.
For more information go to www.ELOCTATE.com or call 1-855-693-5628
Revised September 2017
AHF-WFPLR-WPLR-DEC20